Protein function prediction with Attentive Multimodal Tied Autoencoders.
A PyTorch implementation of Multimodal Tied Autoencoder.
Abstract
A recent method (deepnf) uses a multimodal autoencoder to learn the representation for each protein. The state-of-the-art method has hundreds of millions of parameters to integrate multiple networks. In this project, we propose a multimodal tied autoencoder that constrains the decoder to share parameters with encoders.
Requirements
The codebase is implemented in Python 3.6.9. package versions used for development are just below.
tqdm 4.28.1
numpy 1.15.4
pandas 0.23.4
texttable 1.5.0
scipy 1.1.0
argparse 1.1.0
torch 0.4.1
Datasets
- The interaction networks of Yeast and Human are collected from the STRING database v9.1.
- The functional labels for yeast are obtained from the Munich Information Center for Protein Sequences (MIPS). The functional categories in MIPS are organized in a three-layered hierarchy.
- The functional labels for humans are obtained from the Gene Ontology database. The GO terms for humans are grouped to obtain three distinct levels of functional categories for different specificities.
Preprocessing
To obtain global structure information, we perform random walk with restart followed by Positive Pointwise Mutual Information.
Adjacency matrix A -> Random walk with restart (RWR) -> Positive Pointwise Mutual Information (PPMI) -> feature matrix X
Presenting it as python code:
X = PPMI(RWR(A))
| Network Type | Yeast | Human |
|---|---|---|
| Co-expression | 314,013 | 1,576,332 |
| cooccurence | 2,664 | 36,128 |
| database | 33,486 | 319,004 |
| experimental | 219,995 | 618,574 |
| fusion | 1,361 | 3760 |
| neighborhood | 45,610 | 104958 |
| Dataset | category | # number of labels |
|---|---|---|
| level-1 | 17 | |
| Yeast | level-2 | 74 |
| level-3 | 154 |
| Dataset | category | Biological Process | Cellular Component | Molecular Function |
|---|---|---|---|---|
| 11-30 | 262 | 82 | 153 | |
| Human | 31-100 | 100 | 46 | 72 |
| 101-300 | 28 | 20 | 18 |
Options
Training the model is handled by the main.py script which provides the following command-line arguments.
Input and output options
--data-folder STR The data folder Default is `data/`.
--dataset STR The name of dataset Default is `yeast`.
--annotations-path STR Functional labels. Default is `annotations/`.
--networks-path STR Network datasets. Default is `networks/`.
Model options
--attn-type STR Type of attention Default is `softmax`.
--seed INT Random seed. Default is 42.
--epochs INT Number of training epochs. Default is 20.
--early-stopping INT Early stopping rounds. Default is 10.
--training-size INT Training set size. Default is 1500.
--validation-size INT Validation set size. Default is 500.
--learning-rate FLOAT Adam learning rate. Default is 0.001.
--dropout FLOAT Dropout rate value. Default is 0.5.
--hidden-size INT Layer sizes (Hidden). Default is 2000.
--latent-size INT Layer sizes (LATENT). Default is 600.
--use-cuda BOOL Flag to use cuda Default is False.
Examples
The following commands learn a neural network and score on the test set. Training a model on the default dataset.
python main.py
Training a model for 100 epochs.
python main.py --epochs 100
Increasing the learning rate and dropout.
python main.py --learning-rate 0.1 --dropout 0.9
Training a model with latent-size 64:
python main.py --latent-size 64
Training with Cuda:
python main.py --use-cuda
Results
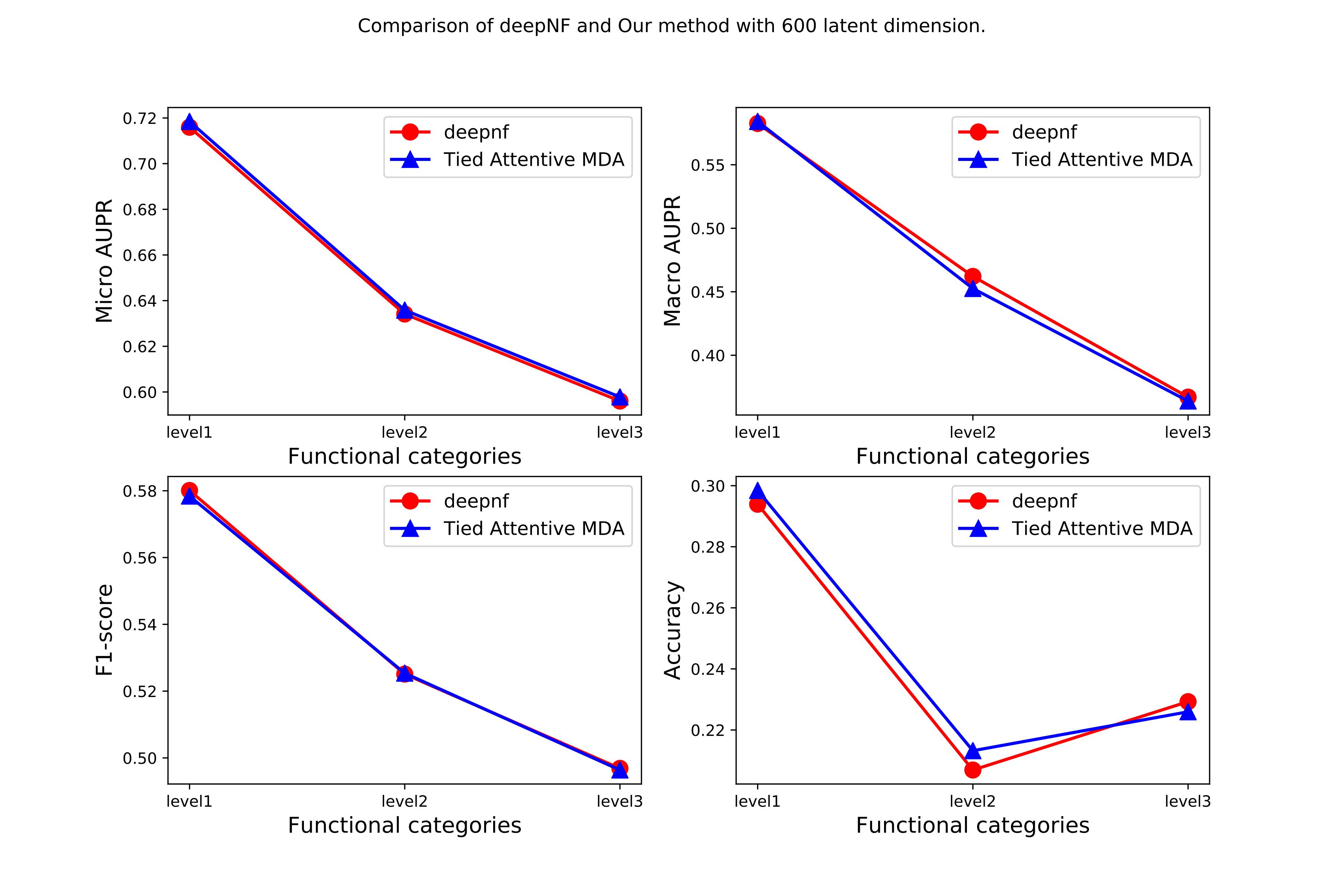
The comparison of the model’s performance with the SOTA method.
| Model | # number of parameters |
|---|---|
| DeepNF | 312 millions |
| TiedDeepNF | 82 millions |
Our model shows a similar performance with 230 million fewer parameters.
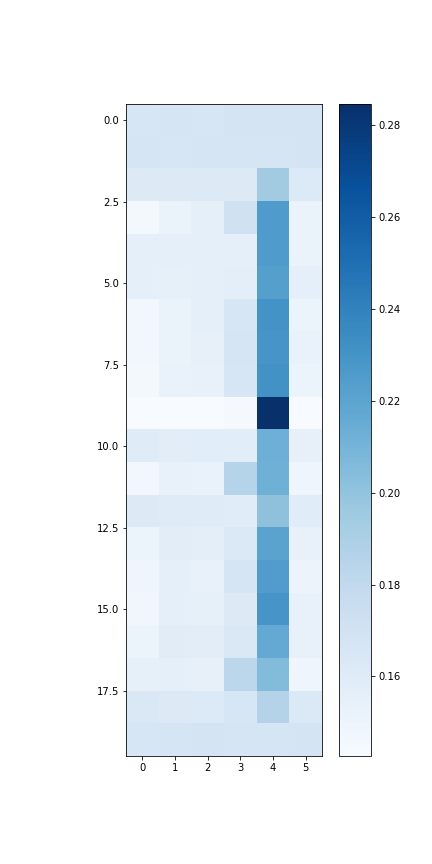
The attention map that indicates the weights given to different networks during feature learning.